
A. Jabbarinick a, A. Houmani a, Z. Harati Zaveh a, B. Abyaz a, A. Ghafari a,c, B. Dashtipour a, M. Sam a,b, F. Asgari Sima a, S. Akbari a,b,c
aNanoSciTec GmbH, Hermann Weinhauser str. 67, Munich, 81867, Germany
bGreen International World Ltd, 128 City Road, London, United Kingdom
c BioMwdEx GmbH, weyringerg 37 Stiege 1, 1040, Wein, Austria
Abstract
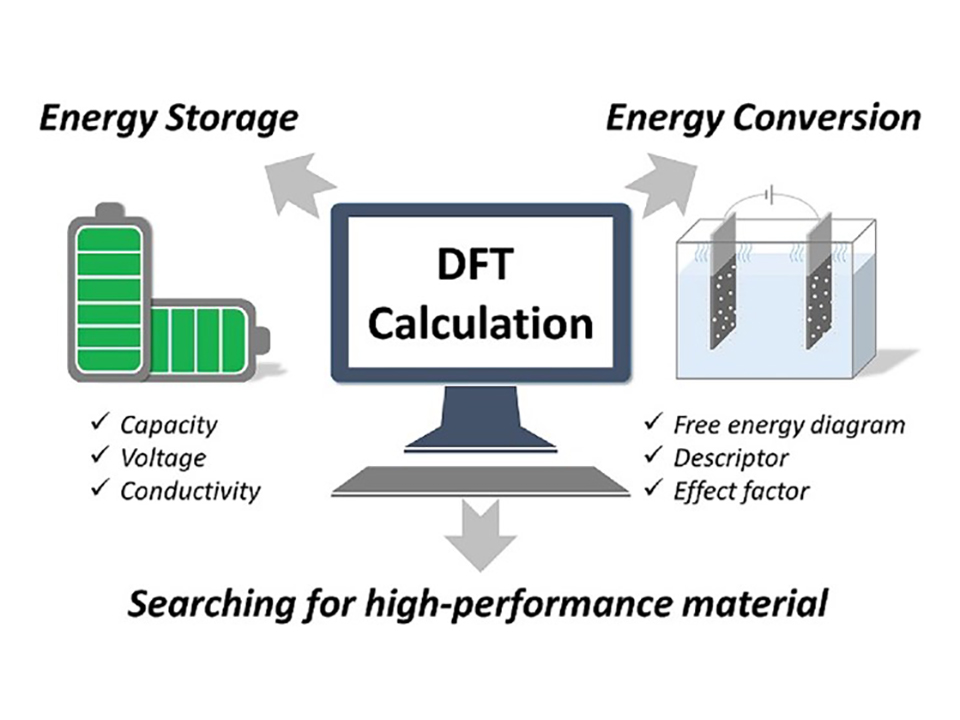
Lithium-ion batteries (LIBs) serve as widespread energy storage solutions in portable electronics, electric vehicles, and renewable energy systems due to their high energy density and rechargeability. Covalent organic frameworks (COFs) exhibit promising potential in LIBs, contributing to enhanced battery performance through improved conductivity, stability, and capacity retention, thus paving the way for more efficient and sustainable energy storage technologies. The advantages of theoretical modeling and simulation methods in COF-based LIBs research are discussed in this contribution. From finite element analysis (FEA) for mechanical perspectives to density functional theory (DFT) for electronic structure considerations and computational fluid dynamics (CFD) for electrolyte and thermal behavior simulations, this study showcases the diverse toolkit used. Electrochemical impedance spectroscopy (EIS) modeling and the integration of machine learning (ML) approaches further enrich the understanding of electrochemical processes and data analysis within lithium batteries. Specific attention is given to modeling and simulating COF-based anodes, cathodes, electrolytes, and separators. This review sheds light on the potential of COFs in revolutionizing lithium battery technology and the importance of computational approaches in advancing their development.
Keywords: Lithium-ion batteries, Covalent organic frameworks, Battery modeling and simulation, Density functional theory, Machine learning
© Article info: Accepted by: 11 January 2024, Published by: 26 January 2024.
Doi Number: https://doi.org/10.52319/j.nanoscitec.2023.28